Chia-Yen Chen, Ruoyu Tian, Tian Ge, Max Lam, Gabriela Sanchez-Andrade, Tarjinder Singh, Lea Urpa, Jimmy Z. Liu, Mark Sanderson, Christine Rowley, Holly Ironfield, Terry Fang, Biogen Biobank Team, The SUPER-Finland study, The Northern Finland Intellectual Disability study, Mark Daly, Aarno Palotie, Ellen A. Tsai, Hailiang Huang, Matthew E. Hurles, Sebastian S. Gerety, Todd Lencz & Heiko Runz
7389 Accesses, 1 Citations, 71 Altmetric
Abstract
Compelling evidence suggests that human cognitive function is strongly influenced by genetics. Here, we conduct a large-scale exome study to examine whether rare protein-coding variants impact cognitive function in the adult population (n = 485,930). We identify eight genes (ADGRB2, KDM5B, GIGYF1, ANKRD12, SLC8A1, RC3H2, CACNA1A and BCAS3) that are associated with adult cognitive function through rare coding variants with large effects. Rare genetic architecture for cognitive function partially overlaps with that of neurodevelopmental disorders. In the case of KDM5B we show how the genetic dosage of one of these genes may determine the variability of cognitive, behavioral and molecular traits in mice and humans. We further provide evidence that rare and common variants overlap in association signals and contribute additively to cognitive function. Our study introduces the relevance of rare coding variants for cognitive function and unveils high-impact monogenic contributions to how cognitive function is distributed in the normal adult population.
Main
Cognitive function is a complex trait consisting of mental processes that include attention, memory, processing speed, spatial ability, language and problem-solving1,2,3,4. General cognitive function and specific cognitive domains can be reliably measured across individuals in the human population and throughout the life span2. Cognitive function in adults, as ascertained either directly via cognitive tests or using proxy measures such as educational attainment (EDU), is strongly influenced by genetics and shows substantial genetic correlation with physical and mental health outcomes as well as mortality1. Nearly 4,000 cognitive function loci of individually small effect sizes have been identified through common variant-based genome-wide association studies (GWAS)2,3,4,5,6. GWAS have also demonstrated shared genetic contributions between cognitive function and neurodevelopmental disorders7,8,9,10, for which large-scale exome studies have identified hundreds of underlying genes7,11,12,13. However, beyond a proposed deleterious effect of exome-wide rare protein-truncating variant (PTV) burden14,15, no studies have yet systematically interrogated the impact of rare coding variants on cognitive phenotypes in the adult general population.
To advance gene discovery for cognitive phenotypes beyond GWAS and gain deeper insights into the shared genetic components between adult cognitive function and neurodevelopmental disorders, we analyzed exome sequencing and genome-wide genotyping data from 454,787 UK Biobank (UKB) participants with measures of cognitive function. We show that adult cognitive function is strongly influenced by the exome-wide burden of rare protein-coding variants and identify and replicate eight genes that are associated with adult cognitive phenotypes. For one of these cognitive function genes, KDM5B, we demonstrate in mice and humans that reduced cognitive function at the population level can be part of a phenotypic spectrum in which cognitive performance depends on the genetic dose of a single gene. Finally, our study bridges a gap between common complex trait and rare disease genetics by demonstrating that adult cognitive function is influenced by additive effects between rare and common variant-based polygenic risk that can be traced to overlapping genomic loci and biological pathways.
Results
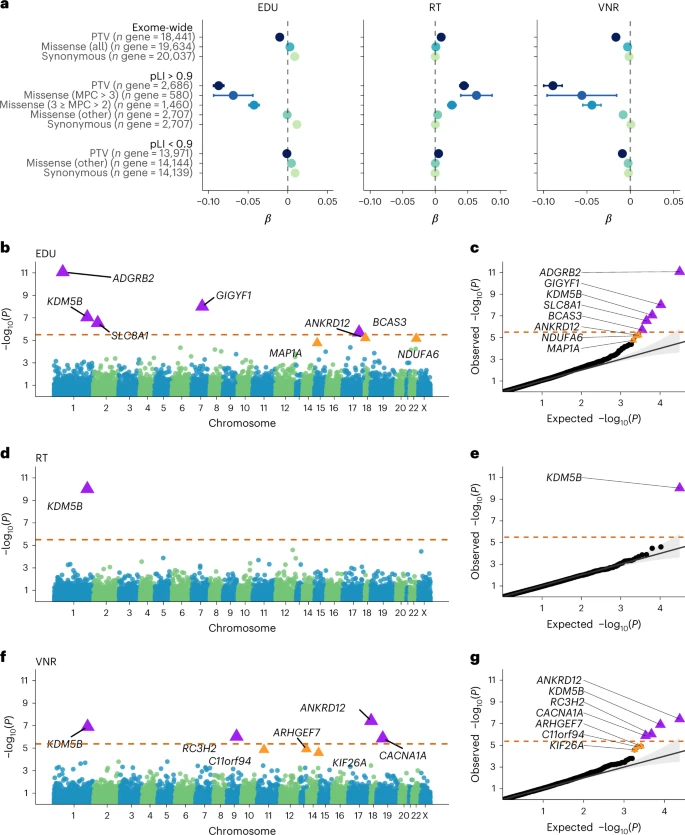
The UKB is a prospective cohort study of over 500,000 participants with extensive health and lifestyle data and genome-wide genotyping and sequencing16,17,18,19,20,21,22. We chose to study the genetic basis of three distinct, yet interrelated phenotypes that previous studies used to approximate adult cognitive function: educational attainment (EDU); reaction time (RT); and verbal-numerical reasoning (VNR)23. EDU is derived from a survey regarding years of schooling, which is genetically correlated with both adult (rg = 0.66) and childhood cognitive function (rg = 0.72) (refs. 5,24,25). RT is based on a digital test that measures processing speed, a component of general cognitive function26,27. VNR is a measure of general cognitive function based on questionnaires. We annotated exome sequencing data from 454,787 UKB participants18,19 for PTVs, missense variants and synonymous variants28,29 and identified rare coding variants with a minor allele frequency (MAF) < 10−5 in the UKB, following previous exome studies on cognition-related traits12,14,15,21,30. We further annotated all variants according to gene intolerance to loss-of-function (LoF) and missense variants for deleteriousness31. In total, we analyzed 649,321 protein-truncating, 5,431,793 missense and 3,060,387 synonymous rare variants.
Rare variants influence adult cognitive function
We first examined the impact of rare coding variant burden on EDU, RT and VNR in unrelated UKB participants of European (EUR) ancestry (n = 321,843; Fig. 1a and Supplementary Tables 1 and 2). We showed that exome-wide PTV and missense burden have significant deleterious effects on cognitive function, which is reflected in lower EDU, longer RT and lower VNR scores per variant count (exome-wide PTV burden: P = 1.95 × 10−21 for EDU, 8.79 × 10−19 for RT and 6.99 × 10−22 for VNR; missense burden: P = 5.95 × 10−24 for EDU, 5.95 × 10−4 for RT and 4.87 × 10−12 for VNR). Consistent with previous exome studies12,13,14,15,30, the most pronounced signals were driven by PTVs and damaging missense variants (missense badness, PolyPhen-2, and constraint (MPC) > 3 and 3 ≥ MPC > 2) in LoF-intolerant genes (pLI ≥ 0.9) (refs. 32,33). The effect sizes of PTV and the MPC > 3 missense burden in LoF-intolerant genes were not significantly different (Fig. 1), suggesting that both classes of variants may impact cognitive function similarly. The synonymous variant burden showed an inverse, albeit small, effect on EDU (exome-wide β = 0.0087, P = 8.59 × 10−75), but not on RT and VNR.
Discussion
In this study, we present a large-scale exome sequencing study on cognitive function phenotypes in the adult general population. Our findings support previous evidence that an increased exome-wide burden of rare PTVs is associated with lower cognitive function14,15 and extend this observation to deleterious missense variants. The large number of exome-sequenced participants in the UKB allowed us to identify eight distinct cognitive function genes, with additional evidence from three independent EUR cohorts. Notably, several of these cognitive function genes have established roles in neurodevelopmental disorders. Our results suggest that a fraction of adults in the normal general population have lower cognitive abilities as a consequence of defects in single disease genes.
Our study is a natural extension of previous GWAS on cognitive function and EDU2,3,4,5,6. While highly successful in identifying associated loci through common variants, applying the GWAS approach to cognitive function has received substantial criticism, especially on potential biases due to ancestry, geography and environmental or cultural differences between subpopulations25,48. Cognitive function is difficult to assess in isolation for its substantial genetic and nongenetic overlap with other traits25. For instance, EDU is not only reflective of childhood and adult IQ, but also strongly correlates with other traits including income, parental age at birth, alcohol dependence or neuroticism25. Furthermore, as suggested by recent studies, EDU is a combination of multiple factors at both phenotypic and genetic levels24,49,50. Using GWAS of EDU and general cognitive function, common genetic associations of EDU have been shown to contain components of both general cognitive ability and noncognitive skills and overlap with psychiatric disorders24,49,50. Nevertheless, we are confident that the results of our exome study are less susceptible to such biases than GWAS. First, we analyzed three distinct phenotypes (EDU, RT and VNR) that each capture different aspects of general cognitive function49. The consistency of exome-wide, gene set-level and gene-level associations across EDU, RT and VNR, which also translate to independent exome-sequenced cohorts, increases the confidence that our gene findings are indeed biologically relevant. Second, five of the eight cognitive function genes (KDM5B (ref. 12,13) (MIM 618109), ANKRD12 (ref. 30), CACNA1A (MIM 183086, MIM 108500 and MIM 617106), GIGYF1 (refs. 12,13) and BCAS3 (MIM 619641)) are either established Mendelian DD genes or have also been identified in previous exome studies on SCZ30,51, DD13 or ASD12. The biological relevance of the genes discovered in this study is consistent with the well-established tight genetic relationships between cognitive traits and diseases. Third, multiple lines of evidence from our analyses yielded clues to a gene’s biological mechanisms and relevance to cognitive function. For instance, ANKRD12 was associated with both EDU and VNR in our exome-wide PTV burden association tests; it is also associated with dysarthria and anarthria, myasthenia gravis and disorders of calcium metabolism in our PTV burden PheWAS. This suggests that cognitive dysfunction in individuals with ANKRD12 LoF is accompanied by imbalances in motor coordination or muscle function and might be part of a yet undescribed genetic syndrome. Likewise, ADGRB2, on top of its association with EDU exome-wide PTV burden analyses, was associated with impairment of cognitive function (ICD-10 code R41.8) in our PTV burden-based PheWAS.
A particularly intriguing example for how diseases with cognitive impairment and adult cognitive function intersect is KDM5B, a gene that encodes a histone lysine demethylase with roles in neuronal differentiation45,52,53,54, which we interrogated further in humans and mice. Biallelic mutations in KDM5B cause an autosomal recessive IDD44,45,53, while HET PTVs have been linked to severe DD7,13,45 and ASD12 with presumed incomplete penetrance45,55. However, because previous studies focused on patients, the relevance of KDM5B LoF in adult cognitive function in the general population has not yet been appreciated. Unlike most other DD genes13, KDM5B is LoF-tolerant, leading to a relatively high PTV carrier rate of approximately 1:1,900 participants in the UKB. As UKB participants tend to be healthier and more educated than the general UK population56,57, it can be expected that KDM5B PTV carrier rates in the general EUR population are even higher. Our results strongly suggest a gene dosage effect for KDM5B, where biallelic, near-complete LoF for KDM5B will lead to more severely impaired cognitive function as observed in patient cohorts, whereas HET KDM5B LoF will present with only moderate cognitive impairment that overlaps with the spectrum of cognitive function in the normal population. Notably, KDM5B showed pleiotropic effects on muscle strength, bone density, growth hormone levels and BD, among others, in our PheWAS. This pleiotropy partially overlaps with the dose-dependent cognitive, behavioral and skeletal symptoms in our Kdm5b mouse model45 as well as KDM5B patients44. It will be interesting to investigate the phenotypic spectrum of KDM5B LoF in humans more comprehensively, for instance, through PheWAS in additional biobanks or targeted follow-up of PTV carriers in recall studies. Genes with a dosage sensitivity like the one described in this study for KDM5B are ideal drug targets because the degree of genetic impairment may guide the development of gene-directed therapeutic interventions58,59. KDM5B is already an established drug target with molecules inhibiting its enzymatic activity in preclinical development for cancer60. It could be interesting to explore whether activators exist that might improve cognitive phenotypes61,62.
A particular strength of exome studies is that genes and variants identified through rare variant tests tend to exhibit much larger effect sizes than common variants identified in GWAS. For example, HET carriers of KDM5B PTVs show on average fewer than 1.51 years of schooling than noncarriers. In contrast, lead SNPs in the most recent EDU GWAS based on three million individuals only show a median 1.4 week increase in schooling per allele (with the 5th and 95th percentiles of the estimated effect being 0.9 and 3.5 weeks, respectively)6. This demonstrates that exome studies may uncover substantially stronger genetic effects and complement GWAS to describe the genetic architecture of cognitive function more comprehensively. This is further supported in the case of ADGRB2 and NDUFA6, which our results suggest as the most probable causal genes in loci identified in EDU and cognitive function GWAS5.
With both exome sequencing and genome-wide genotype data in the UKB, we were able to explore the relative contribution of common variant-based polygenic risk and rare coding variant burden to cognitive function. Our results provide evidence that rare coding variants affect EDU and VNR additively to PRS and thus suggest that genetic prediction can be further improved by combining PRS and the rare coding variant burden. Similar findings were reported previously for other common complex phenotypes51,63,64,65. Although the phenotypic variance explained by rare coding variants is much smaller than that explained by PRS because of allele frequency constraints, rare coding variants provide orthogonal predictive power that is not relying on external training GWAS (like PRS) and is thus less susceptible to biases66.
Future studies are needed to better understand the biological basis of how the genes and variants reported in this study impact cognitive function and related diseases. Moreover, our findings do not imply direct applications in clinical practice, such as for prenatal genetic screening67,68, and should be interpreted with similar caution as reported in GWAS6. Further work is also needed to assess how well the results from our study can be extrapolated to ancestries other than EUR populations. Nevertheless, our results provide a starting point toward expanding our knowledge on how rare genetic variants impact cognitive function at the population level and support a convergence of rare and common genetic variations that jointly contribute to the spectrum of cognitive traits and diseases.
Methods
The UKB is approved by the North West Multi-centre Research Ethics Committee (https://www.ukbiobank.ac.uk/learn-more-about-uk-biobank/about-us/ethics). The current study was conducted under UKB application no. 26041. The data in the UKB were collected after written informed consent was obtained from all participants. The Human Research Committee of the MGB approved the Biobank research protocol (no. 2009P002312) (ref. 69). The data in the MGBB were collected after broad-based written consent was obtained from all participants. The Coordinating Ethical Committee of the Helsinki and Uusimaa Hospital Region approved the SUPER-Finland study on 16 July 2015 (pilot) and 9 February 2016 (full study). All participants of the SUPER-Finland study signed an informed consent that permits research use of collected samples and data. The ethical committees of the Northern Ostrobothnia Hospital District and the Hospital District of Helsinki and Uusimaa approved the NFID study. All participants or their legal guardians provided written informed consent to participate in the study. The breeding and housing of mice, and all procedures for the Kdm5b LOF mouse experiments were assessed by the Animal Welfare and Ethical Review Body of the Wellcome Sanger Institute and conducted under a UK Home Office license (no. P6320B89B) in accordance with institutional guidelines.
Cognitive function phenotypes in UKB
The UKB is a prospective cohort study of the UK population with over 500,000 participants16. Participants were aged between 40 and 69 years at recruitment in 2006–2010 and provided extensive phenotypic data17. We extracted three cognitive function phenotypes for our analysis: EDU; RT; and VNR23. EDU is based on a survey of years of schooling that reflects both cognitive function and noncognitive components24. We extracted the UKB data field 6138 ‘Qualifications’ collected at baseline as a measure of EDU and converted multiple-choice categories to years of schooling as outlined by Lee et al.5. RT is a measure of processing speed, which is a component of general cognitive function2,4,26,27, implemented as a digital assessment at baseline (UKB data field no. 20023) (ref. 2). VNR is a score measured using a structured questionnaire, which contains 13 questions that focus on assessing crystalized and fluid intelligence in both verbal and numerical aspects (UKB data field no. 20016). Note that only a subset of 165,453 UKB participants completed the VNR assessment at baseline, while EDU (n = 497,844 at baseline collection) and RT (n = 496,660 at baseline collection) data were collected for almost the entire UKB. For the association analyses in the UKB samples, we rank-based inverse-normal transformed the phenotypes. While higher EDU and VNR scores indicate better cognitive function, longer RT represents worse cognitive function.
The UKB whole-exome sequencing data
Whole-exome sequencing (WES) data from UKB participants was generated by the Regeneron Genetics Center on behalf of the UKB Exome Sequencing Consortium, which is a collaboration between AbbVie, Alnylam Pharmaceuticals, AstraZeneca, Biogen, Bristol Myers Squibb, Pfizer, Regeneron and Takeda18. Briefly, WES was done on an Illumina NovaSeq 6000 platform using xGen Exome capture kits. Sequencing reads were aligned to the GRCh38 reference genome using the Burrows–Wheeler Aligner-MEM (v.0.7.17) (refs. 18,70). Single-nucleotide variants and indels were called by first generating gVCF files using the WeCall variant caller v.1.1.2 (Genomics PLC) and then joint-called using the GLnexus joint genotyping tool (v.0.4.0) (refs. 18,21,71). The joint-called, project-level VCF was then filtered by the Regeneron Genetics Center quality control (QC) pipeline (the ‘Goldilocks’ set). As of November 2020, we obtained QC-passed WES data from 454,787 UKB participants. The UKB can release these data publicly to approved researchers via their Research Analysis Platform.
We annotated variants using Variant Effect Predictor (VEP) v.96 (ref. 28) with genome build GRCh38. Stop-gain, splice site-disruptive and frameshift variants were further assessed by Loss-Of-Function Transcript Effect Estimator (LOFTEE) (a VEP plugin)29 and high-confidence predicted LOF variants were retained for analysis. Missense variants were further annotated for deleteriousness using the MPC score31. We also annotated variants based on gene intolerance to LOF using pLI (probability of being LOF-intolerant) v.2.1.1 (refs. 29,32). All predicted variants were mapped to GENCODE72 (release 30) canonical transcripts.
For the association analysis, we filtered variants to include only those with a MAF < 1.0 × 10−5 in the UKB (649,321 PTVs, 5,431,793 missense variants and 3,060,387 synonymous variants) to enrich for pathogenic variants. In previous exome studies, the impact of an exome-wide ultrarare variant burden was associated with EDU, ID and psychiatric disorders. In these studies, ultrarare variants were defined as variants observed in fewer than 1 in 74,839 individuals (allele frequency < 1.34 × 10−5) or 1 in 201,176 individuals (allele frequency < 2.49 × 10−6) (refs. 14,15) in external reference samples. A recent large-scale exome study for SCZ also adopted a minor allele count cutoff of fewer than 5 alleles in 24,248 cases and 97,322 population controls, which corresponds to a MAF cutoff of 2.06 × 10−5 (ref. 30). Our choice of variant filtering for MAF < 1.0 × 10−5 is in line with these previous studies.
The UKB genome-wide genotype data
We used imputed genotype data provided by the UKB with additional QC filtering. Genome-wide genotyping was performed for all UKB participants and imputed with the Haplotype Reference Consortium73 and UK10K74 plus 1000 Genomes Project reference panels75, resulting in a total of more than 90 million variants. We performed QC on the genotyping data by filtering out variants with an imputation quality INFO score < 0.8 and variants with a MAF < 0.01 using PLINK v.2.00 (ref. 76). We filtered out 1,804 individuals whose reported gender differed from their genetic gender, individuals showing sex chromosome aneuploidies, as well as 133 individuals who had withdrawn from the UKB (as of 24 August 2020).
To identify UKB samples from different genetic ancestries, we performed population assignment based on population structure using principal component analysis (PCA) with the 1000 Genomes Project reference samples (n sample = 2,504) from five major population groups: AFR; American (AMR); East Asian (EAS); EUR; and SAS. Details of the genetic PCA-based population assignment can be found in the Supplementary Information. We identified 8,406 AFR samples, 1,085 AMR samples, 1,609 EAS samples, 458,197 EUR samples, 9,224 SAS samples and 8,874 samples without explicit population assignment. Due to the small sample sizes, we did not analyze further the samples in the EAS and AMR groups. We also did not analyze further samples without an explicit population assignment. Within-population PCA was performed for the AFR, EUR and SAS samples for subsequent association analyses.
Gene set-based rare coding variant burden test
Analysis overview
To estimate the association between cognitive function phenotypes (EDU, RT and VNR) and gene set-based rare coding variant burdens, we rank-based inverse-normal transformed the phenotypes and fitted a linear regression in unrelated UKB samples in samples from the same population group (as described in the section on population assignment). To minimize potential population stratification and confounding, we adjusted for sex, age, age2, sex by age interaction, sex by age2 interaction, top 20 principal components (PCs) and recruitment centers (as categorical variables) in all association analyses. We ran additional sensitivity analyses accounting for 40 PCs to assess the potential residual population stratification and found that the exome-wide burden results were consistent (Supplementary Table 2). The effect size (β), 95% CIs and P values were calculated for each burden association. The significance level of the burden association was determined using Bonferroni correction for the number of association tests in the defined set of analysis and is provided in Supplementary Tables 1, 2, 6, 7, 10 and 18–20.
Exome-wide burden
To characterize the effects of exome-wide rare coding variant burden on cognitive function, we calculated the cumulative minor allele counts of rare coding variants (MAF < 1.0 × 10−5) for each variant functional class as defined by the VEP28, LOFTEE32, MPC31 and pLI scores32. We defined the following variant classes: PTVs; high-confidence LOF variants; missense variants classified according to deleteriousness (MPC) into tier 1 for MPC > 3, tier 2 for 3 ≥ MPC > 2 and tier 3 for other missense variants not in the previous two tiers; and synonymous variants (identified by VEP). We further classified variants according to the LOF intolerance of the gene (pLI ≥ 0.9 or pLI < 0.9) in which the variant resides. The exome-wide rare coding variant burdens for each variant class were calculated and burden association tests were performed in the EUR, SAS and AFR samples in the UKB.
Gene set burden
We also calculated the rare coding variant burdens for several gene sets, including genes identified in: (1) exome studies for ASD (n gene = 102)11, DD (n gene = 285)13 and the DDG2P (https://www.deciphergenomics.org/ddd/ddgenes; n gene = 2,020); (2) GWAS for EDU (n gene = 1,140) (ref. 5), cognitive function (n gene = 807) (ref. 4), SCZ (n gene = 3,542) (ref. 77), BD (n gene = 218) (ref. 78) and depression (n gene = 269) (ref. 79); (3) gene sets annotated in the Molecular Signatures Database (v.7.2; n gene set annotated = 13,011); (4) gene sets with brain tissue expression specificity annotated in the Human Brain Atlas80 (n gene annotated = 16,270). Details on the calculation of gene set burdens and association analyses can be found in Supplementary Information.
Exome-wide, gene-based PTV burden test
To identify genes associated with adult cognitive function, we calculated the rare PTV burden for each gene and performed burden association analyses. We used two-step whole-genome regression implemented in regenie for association testing34. Regenie accounts for population stratification and sample relatedness, which allowed us to leverage a larger sample size by including related samples. Regenie first fits a stacked block ridge regression to obtain leave-one-chromosome-out (LOCO) genetic prediction of the phenotype of interest; in the second step, the association test is carried out by fitting regression models conditioning on the LOCO predictions derived in the first step.
For the regenie step 1 regression, we first performed sample QC and then genotype QC by excluding variants with a genotyping call rate less than 90%, Hardy–Weinberg equilibrium test P < 10−15 and MAF < 1%. This retained 565,124 genotyped variants for the step 1 regression. We fitted a regenie first-step regression for rank-based inverse-normal transformed EDU, RT and VNR separately, adjusting for sex, age, age2, sex by age interaction, sex by age2 interaction, top 20 PCs and recruitment centers with tenfold cross-validation (regenie v.1.0.6.7) (ref. 34). For the regenie step 2 association test, we implemented an in-house pipeline (R v.3.6.1) for rare PTV burden association tests conditioned on the first-step LOCO prediction, following the linear regression model for association testing described in the regenie publication34. We treated the LOCO prediction as an offset in the linear regression model where rank-based inverse-normal transformed EDU, RT and VNR were regressed on gene-based rare PTV burden, adjusting for the same covariates used in the step 1 regression. We excluded genes with fewer than ten PTV carriers from the gene-based PTV burden analysis, which leads to a variable number of tests performed for each phenotype, especially for VNR, which has a much smaller sample size. We repeated the two-step regenie regression in the UKB EUR, SAS and AFR samples.
For the EUR samples, the sample sizes and number of genes tested for each cognitive function phenotype were as follows: n sample = 393,758 and test n = 15,782 for EDU; n sample = 394,600 and test n = 15,798 for RT; and n sample = 159,026 and test n = 11,905 for VNR. The Bonferroni correction for multiple testing was based on the actual number of tests performed per phenotype and across the three phenotypes. The significance levels for the gene-based rare PTV burden association tests were Bonferroni-corrected for the number of tests for each phenotype separately, which are 0.05/15,782 = 3.17 × 10−6 for EDU, 0.05/15,798 = 3.16 × 10−6 for RT and 0.05/11,905 = 4.20 × 10−6 for VNR. Note that seven of the eight cognitive function genes (that is, all except BCAS3) identified in our PTV burden association analysis in the UKB EUR samples were also exome-wide-significant after Bonferroni correction across all tests (P < 0.05/43,485 = 1.15 × 10−6). Additionally, we identified five genes with an FDR Q < 0.05 for EDU and VNR in the UKB EUR samples. For the SAS samples, the sample sizes and number of genes tested were as follows: n sample = 8,181 and test n = 1,247 for EDU; n sample = 8,018 and test n = 1,187 for RT; n sample = 4,430 and test n = 331 for VNR. For the AFR samples, the sample sizes and number of genes tested were as follows: n sample = 7,504 and test n = 887 for EDU; n sample = 7,331 and test n = 844 for RT; n sample = 3,890 and test n = 179 for VNR.
Replication cohorts
To replicate our gene findings from the exome-wide, gene-based PTV burden tests in the UKB, we performed gene set-based and gene-based PTV burden association tests in three independent cohorts with samples of EUR ancestry: the SUPER-Finland study; the NFID study; and the MGBB. Details of phenotype, genotype and exome sequencing data processing and QC can be found in the Supplementary Information. In each replication cohort, we calculated PTV burdens for two cognitive function gene sets, including the eight genes with Bonferroni-corrected significance and the 13 genes with FDR significance identified in the UKB EUR samples. We also calculated the PTV burdens of individual cognitive genes with at least five rare PTV carriers (ADGRB2, KDM5B, GIGYF1, ANKRD12 and KIF26A) in the NFID study. PTV burden association tests were then performed between the PTV burdens and cognitive traits in the replication cohorts. For the SUPER-Finland study, association tests were performed between PTV burdens and DD/ID, academic performance compared with schoolmates and EDU using either linear or logistic regression, adjusted for ten PCs, imputed sex, sequence assay and total number of coding variants in the genome. For the NFID study, we tested associations between PTV burdens and DD/ID using a logistic regression, adjusted for sex and the top ten PCs. In addition, we performed a meta-analysis of the DD/ID association with PTV burdens between the SUPER-Finland and NFID studies using an inverse-variance weighted random-effects meta-analysis. For the MGBB, we tested the association between PTV burdens and EDU using a linear regression, which was adjusted for sex, age, age2, sex by age interaction, sex by age2 interaction and the top 20 PCs. ORs, 95% CIs and P values were calculated for all association tests and meta-analyses.
Phenome-wide association analysis
To explore the cognitive function genes identified for potential pleiotropic effects, we performed a PTV burden phenome-wide association analysis across 3,150 UKB phenotypes derived semiautomatically. Binary phenotypes included ICD-10 codes from inpatient records (congenital malformations; deformations and chromosomal abnormalities; diseases of the circulatory system; diseases of the digestive system; diseases of the eye and adnexa; diseases of the genitourinary system; diseases of the musculoskeletal system and connective tissue; diseases of the nervous system; diseases of the respiratory system; diseases of the skin and subcutaneous tissue; endocrine, nutritional and metabolic diseases; mental, behavioral and neurodevelopmental disorders; neoplasms; pregnancy; childbirth and the puerperium; symptoms, signs and abnormal clinical and laboratory findings; not elsewhere classified) and death records (ICD-10 cause of death), self-reported illness (cancer, non-cancer), self-reported medication, surgery and operation codes, and family history (father’s, mother’s and siblings’ illnesses were combined into a single phenotype for each of the 12 family history illnesses ascertained in the UKB questionnaires). Quantitative phenotypes included biomarkers such as blood cell count, blood biochemistry, infectious disease antigen assays and physical measurements. A list of all phenotypes with phenotype categories, UKB field numbers and phenotype full names can be found in Supplementary Table 9.
We restricted the phenome-wide association analysis to 321,843 unrelated UKB EUR samples and excluded binary phenotypes with fewer than 100 cases in our analysis. PTV burden testing for binary phenotypes was performed in all individuals using logistic regression, controlling for sex, age, age2, sex by age interaction, sex by age2 interaction, top 20 PCs and assessment centers. For binary phenotypes with a PTV burden association P < 0.01, we repeated the analysis using Firth’s logistic regression to account for situations where the logistic regression outputs may be biased due to separation81. For quantitative phenotypes, we excluded phenotypes with fewer than 100 observations. For each quantitative phenotype, individuals with outlier phenotype values (>5 s.d. from the mean) were excluded. The PTV burden test for quantitative traits was performed using linear regression on rank-based inverse-normal transformed phenotypes in all individuals, controlling for sex, age, age2, sex by age, sex by age2, top 20 PCs and assessment centers. We defined a Bonferroni-corrected phenome-wide significance threshold (using the number of tests per gene) of 1.59 × 10−5 (0.05/3,150).
Characterization of cognitive phenotypes in KDM5B and CACNA1A PTV carriers
KDM5B is an established Mendelian disease gene, with HOM or compound HET mutations causing autosomal recessive ID (MIM 618109) (refs. 44,45). Similarly, CACNA1A is also an established disease gene with HET mutations causing developmental and epileptic encephalopathy (MIM 617106) (refs. 41,42), type 2 episodic ataxia (MIM 108500) (ref. 40) or spinocerebellar ataxia (MIM 183086) (refs. 38,39). To better understand the relationship between PTVs in KDM5B and CACNA1A and cognitive function phenotypes, we first processed EDU and VNR in the UKB EUR samples by residualizing EDU and VNR with sex, age, age2, sex by age interaction, sex by age2 interaction, top 20 PCs and recruitment centers and then standardized the residuals using rank-based inverse-normal transformation. Then, we plotted the standardized, residualized EDU and VNR for each PTV carrier against the genomic position of the PTV to characterize the phenotypic distribution of KDM5B and CACNA1A PTV carriers. We further compared the standardized, residualized EDU and VNR between three groups of PTV carriers for KDM5B and CACNA1A: (1) PTV carriers who do not have any inpatient ICD-10 diagnostic codes for neurological, psychiatric or neurodegenerative disorders or carry ClinVar pathogenic or likely pathogenic variants; (2) PTV carriers with inpatient ICD-10 diagnostic codes for neurological, psychiatric or neurodegenerative disorders; (3) PTV carriers of ClinVar pathogenic or likely pathogenic variants.
Kdm5b mouse model
To experimentally investigate the potential additive dosage effect of Kdm5b LoF, we performed behavioral tests, morphological measurements and brain differential gene expression analysis in WT, HET and HOM Kdm5b LoF mice. A mouse Kdm5b LoF allele (Mouse Genome Informatics ID: 6153378) was generated previously45 using CRISPR/CAS9 mediated deletion of coding exon 7 (ENSMUSE00001331577), leading to premature translational termination due to a downstream frameshift. Breeding of testing cohorts was performed on a C57BL/6NJ background. Mice were housed in specific pathogen‐free mouse facilities with a 12-hour light–dark cycle (lights on at 7:30), an ambient room temperature of 21 °C and 55% humidity at the Research Support Facility of the Wellcome Sanger Institute. They were in mixed genotype cages (2–5 mice), and housed in individually ventilated cages (GM500, Tecniplast) containing Aspen chip bedding and environmental enrichment (Nestlets nesting material and cardboard play tunnels, Datesand). Food and water were provided ad libitum.
We applied a battery of behavioral tests commonly used to study mice for signs of perturbed neurodevelopment, including light–dark box (adapted from Gapp et al.82), Barnes maze probe trial and new object recognition (Supplementary Information). We assessed a cohort of 25 WT, 34 HET and 15 HOM Kdm5b mutant male mice at 10 weeks of age. Behavioral tests were carried out between 9:00 and 17:00, after 1 hour of habituation to the testing room. Experimenters were blind to genotype; mouse movements were recorded with an overhead infrared video camera for later tracking using automated video tracking (EthoVision XT 11.5, Noldus Information Technology). We also measured mouse cranial length and width, skull height and transitional vertebrae phenotype with X-ray whole-body radiography for 15 Kdm5b+/+, 12 Kdm5b+/− and 9 Kdm5b−/− mice (Supplementary Information).
All statistical analyses of mouse data were performed using R (v.4.1.3). Data were first transformed to achieve normality, using Box–Cox transformation (MASS package v.7.3–55) for behavioral data (λ limit = −2, 2) or quantile normalization (qnorm function) for X-ray data. Testing for genotype effect was performed using a double generalized linear model (dglm package v.1.8.5). The type of object used for new object recognition had a small (6%) and significant (P = 0.036) effect; therefore, it was used as a covariate for Box–Cox transformation and dglm. For visualization purposes, residual values were calculated from the linear model and z-scores were calculated relative to WT.
We also performed differential gene expression analysis for the Kdm5b mouse to assess the impact of the Kdm5b HET and HOM mutations on brain gene expression. RNA-seq was done for whole-brain embryonic tissue, and for the FC, HIP and CB of adult WT, HET and HOM Kdm5b mice. Differential gene expression and log2 fold changes were obtained, and P values for differences in gene expression were calculated. A P threshold of 0.10 was used to identify significant differences between WT and mutant samples. In addition, Gene Ontology (GO) enrichment analysis was performed to identify functionally enriched terms in the DEGs (with a 5% FDR threshold). In all analyses, the background consisted of only genes expressed in the tissue studied. GO terms with more than 1,000 genes were excluded from the analysis. The European Nucleotide Archive (ENA) accession numbers for the RNA-seq sequences reported are listed in Supplementary Table 17. Further details on Kdm5b mouse RNA extraction, sequencing, data processing and analyses can be found in the Supplementary Information.
Overlapping rare and common variant association signals
To compare and contrast the genetic loci identified through the common variant association tests in GWAS to the genes identified in our rare PTV burden analysis, we cross-checked all independent genome-wide significant variants in the most recent, largest EDU GWAS5 and cognitive function GWAS4 with the 13 cognitive function-associated genes identified in the current study. For the EDU GWAS, we assessed the independent genome-wide significant variants listed in Lee et al.5 (See Supplementary Table 2 for any nearby genes with significant PTV burden association with cognitive function phenotypes.) We identified one SNP, rs10798888 (chr1:31733498 (GRCh38); MAF = 0.1725; EDU association P = 5.15 × 10−14), where ADGRB2 (PTV burden P = 8.55 × 10−12 for EDU in the UKB EUR samples) is located in a nearby region. We then extracted the region surrounding SNP rs10798888 from the full summary statistics of the EDU GWAS (excluding the 23andMe data) obtained from the Social Science Genetic Association Consortium (SSGAC), generated regional plots (https://my.locuszoom.org/)83 of the GWAS results and compared these with the cognitive function phenotypes (EDU and VNR) among PTV carriers in the UKB EUR samples. Variants in the GWAS regional plots were further annotated for previous GWAS associations registered in the GWAS catalog using LocusZoom’s automated annotation feature.
For the cognitive function GWAS, we processed the GWAS summary statistics from Lam et al.4 with a GWAS summary statistics QC pipeline50 and used FUMA84 to identify independent genome-wide-significant loci for cognitive function from the GWAS. We identified one genome-wide-significant locus with the top independent genome-wide-significant SNP rs5751191 (chr22:41974987 (GRCh38), association P = 2.02 × 10−12) that overlapped with NDUFA6 (PTV burden P = 6.98 × 10−6, FDR Q = 0.016 for EDU in the UKB EUR samples). We extracted variants in the region that covered the variants in linkage disequilibrium with the top SNP rs5751191 (R2 > 0.6) to generate a regional plot and identified genes in the region to extract the corresponding PTV burden association P values and the number of PTV carriers in the UKB EUR samples.
Contributions of common variants and rare damaging coding variants to EDU and VNR
We examined the relative contribution of common variants and rare damaging coding variants to cognitive function. To do so, we first calculated the PRS to capture the impact of genome-wide common variants on cognitive function, using imputed genome-wide genotypes and variant weights derived using PRS-CS47 based on a cognitive function GWAS meta-analysis4 and a precomputed linkage disequilibrium reference panel based on the 1000 Genomes Project phase 3 EUR superpopulation samples. The cognitive function GWAS meta-analysis included only samples of EUR ancestry from the latest cognitive genomics consortium (COGENT) data freeze, excluding samples from the UKB4. The PRS-CS global shrinkage parameter ϕ was set to 0.01 because cognitive function is highly polygenic4. Using PRS-CS-derived variant weights and QC-imputed genotypes, we calculated PRS as a weighted sum of counted alleles across the genome using PLINK v.2.00. Then, after the exome-wide burden analysis, we identified rare damaging coding variant carriers as carriers of rare PTV or damaging missense variants with an MPC > 2 in LOF-intolerant genes (pLI > 0.9) across the exome.
To demonstrate the relative impact of PRS and rare damaging coding variant carrier status, we plotted standardized, residualized EDU and VNR against PRS, stratified according to rare damaging coding variant carrier status in unrelated UKB EUR samples. The phenotypes were residualized by sex, age, age2, sex by age interaction, sex by age2 interaction, top 20 PCs and recruitment centers and then rank-based inverse-normal transformed. The samples were grouped by PRS in 20% or 2% quantiles and are shown in Fig. 5 and Extended Data Fig. 9. The median of standardized, residualized EDU and VNR was calculated and plotted for each PRS group. We further assessed the prediction performance of cognitive function PRS and rare damaging coding variant carrier status for EDU and VNR. We fitted linear regression models by regressing rank-based inverse-normal transformed EDU and VNR on PRS and rare damaging coding variant carrier status jointly, adjusted for covariates, in unrelated UKB EUR samples. The regression coefficients, association P values and partial R2 were estimated85. We further examined the interaction between PRS and rare damaging coding variant carrier status by adding an interaction term to the linear regression model and tested for significant interaction effects. We also modeled PRS as both a continuous and a binary variable by dividing samples in the top 10% PRS group versus the remaining 90%.